![]()
![]()
![]()
Глава II.6.1.
Молекулярная
механика (ММ)
Выбор в меню Setup пункта, соответствующего молекулярной механики, позволяет использовать классический Ньютоновский метод вычислений энергии одной точки, равновесной геометрии и молекулярной динамики объектов вместо квантово-механического подхода (одного из полуэмпирических методов или неэмпирического метода Хартри-Фока (ab initio)).
В методе молекулярной механики атомы рассматриваются как ньютоновские частицы, которые взаимодействуют друг с другом посредством неких потенциальных полей, задаваемых эмпирически. Потенциальная энергия взаимодействия зависит от длины связей, углов связи, торсионных углов и нековалентных взаимодействий (в т.ч. сил Ван-дер-Ваальса, электростатических взаимодействий и водородных связей). В этих расчетах силы, действующие на атомы, представляются в виде функций координат атомов.
Примечание: Если в рабочей области выделена только часть системы, в расчет будут включаться взаимодействия только выделенной части. При оптимизации геометрии и расчетах методом молекулярной динамики, в этом случае, только атомы выделенной части будут менять свое положение в пространстве, тогда как невыделенные – нет, при этом в расчетах будет учитываться потенциальные взаимодействия между частями системы.
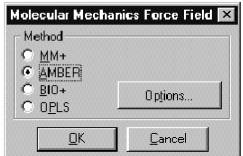
Для начала расчетов методом молекулярной механики в диалоговом окне необходимо выбрать Force fild (Силовое поле) - потенциальную функцию для расчетов. Можно выбрать один из четырех методов (MM+, AMBER, BIO+, OPLS), ссылки на которые можно увидеть в диалоговом окне.
Метод MM+ разрабатывался для органических молекул. Он учитывает потенциальные поля, формируемыми всеми атомами рассчитываемой системы и позволяет гибко модифицировать параметры расчета в зависимости от конкретной задачи, что делает его, с одной стороны, наиболее общим, а с другой – резко увеличивает необходимые ресурсы по сравнению с другими методами молекулярной механики. Ряд возможностей для изменения параметров этого метода можно получить, выбрав кнопку Options в пункте выбора Силового поля.
Метод AMBER разрабатывался для белков и нуклеиновых кислот. В нем существует возможность выбрать опцию либо учета всех атомов по отдельности, либо опцию объединенного атома, под которым подразумевается группа эквивалентных атомов с одинаковыми свойствами. В последнем случае несколько атомов, либо их групп, обрабатываются как один атом с одним типом.
BIO+ разрабатывался для биологических макромолекул и во многом повторяет AMBER.
OPLS разработан для белков и нуклеиновых кислот. Он подобен AMBER, но более точно обрабатывает нековалентные взаимодействия.
Диалоговое окно
молекулярной механики MM+ Options
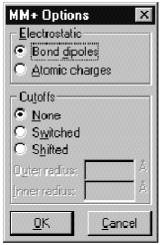
Диалоговое
окно ММ+ содержит набор настроек для соответствующего силового поля.
Electrostatics (Электростатика) Нековалентные электростатические взаимодействия рассчитываются с использованием взаимодействий дипольного типа или частичных атомных зарядов.
Ö Bond dipoles используется для расчетов нековалентных электростатических взаимодействий. Значение этого параметра определяется в файле параметров MM+.
Ö Atomic charges используется для расчетов нековалентных электростатических взаимодействий. Вы можете задавать неполные (частичные) атомные заряды посредством меню Build, пункта Set Charge или Вы можете проводить полуэмпирические или ab initio расчеты, сначала рассчитывая частичные заряды для каждого атома методом Мулликена.
Cutoffs (Отключение) этот параметр определяет минимальное расстояние для нековалентных взаимодействий.
Ö Switched вводит сглаживающую функцию при расчетах молекул в Periodic Box (Периодический ящик). Этот подход позволяет плавно уменьшать слабые взаимодействия вплоть до нуля, перемещаясь из внутренней сферы во внешнюю. В этом случае HyperChem устанавливает параметр Switched и значения внутренней (Inner) и внешней (Outer) сфер (Spheres).
Ö None. Этот параметр устанавливается для расчета систем в вакууме.
Ö Shifted вводит сглаживающую функцию, которая действует на все пространство от 0 до внешней сферы. Эта функция позволяет плавно уменьшать нековалентные взаимодействия до 0.
Ö Outer radius для параметров Switched и Shifted определяет минимальное расстояние, на котором нековалентные взаимодействия становятся равными 0. Обыкновенно это значение выбирается не менее чем на 4 ангстрема больше, нежели чем внутренний радиус. Для периодических граничных условий это значение равно половине минимального размера периодического ящика.
Ö Inner radius выбирается только в случае установки Switched cutoffs. Это максимальное межатомное расстояние для полного учета нековалентных взаимодействий. В случае выбора периодических граничных условий это значение выбирается на 4 ангстрема меньше, нежели чем половина минимального размера Периодического ящика, или менее, вплоть до 0. Внимание, установки Cutoffs возвращаются к своим стандартным значениям в случае, когда в рабочее поле помещается новая молекула.
Диалоговое
окно опций силового поля (Force Field Options Dialog Box)
Это окно используется для выбора параметров силовых полей AMBER, BIO+ и OPLS. HyperChem хранит значения этих параметров, исключая параметры Cutoffs, в Registry или в файле chem..ini и использует их для последующих вычислений.
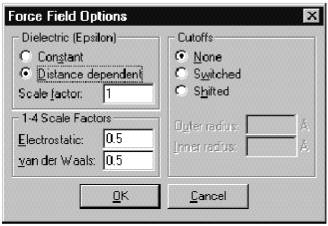
Dielectric permittivity (epsilon) (диэликтрическая постоянная). Параметры Constant (Постоянная) или Distance dependent (Зависящая от расстояния) определяют методы расчета диэлектрической постоянной эпсилон, фактора, который модифицирует взаимодействие зарядов (и электростатического потенциала).
Ö Constant (Постоянная). Выбор этого параметра делает диэлектрическую постоянную константой и соответствует периодическим граничным условиям Периодического ящика. Выбор этого пункта соответствует веществу, находящемуся в газовой фазе, либо в идеальном растворе.
Ö Distance dependent (Зависящая от расстояния). Выбор этого параметра делает эпсилон пропорциональной межатомному расстоянию. Подобный подход аппроксимирует эффект сольватации в отсутствии идеального растворителя и позволяет убыстрять расчеты. Данный параметр рекомендуется использовать при расчетах методом OPLS. Так как данный параметр моделирует присутствие сольвента, его не следует применять, когда молекулы сольвента присутствуют в моделируемой системе.
В случае выбора параметра Constant эпсилон (epsilon)=(диэлектрическая постоянная свободного пространства) * (масштабный множитель (Scale factor)). В случае выбора параметра Distance dependent эпсилон (epsilon)=(диэлектрическая постоянная свободного пространства) * (масштабный множитель (Scale factor)) * (межатомное расстояние). Масштабный множитель должен быть >=1. По умолчанию он принимается равным 1, что удовлетворяет для большинства рассчитываемых систем.
1–4 Scale factor (Масштабный множитель 1-4) нековалентные взаимодействия между атомами, разделенными в точности тремя связями, умножаются на этот множитель.
Ö Electrostatic (Электростатика) модифицирует силу взаимодействия зарядов между атомами, разделенными тремя связями. Этот параметр меняется в пределах от 0 до 1. Для силового поля AMBER и OPLS необходимо использовать 0.5, для BIO+ рекомендуется 1.0, 0.5 или 0.4 в зависимости от набора других параметров.
Ö Van-der-Waals (Ван-дер-Ваальс) модифицирует ван-дер-ваальсовы взаимодействия между атомами, разделенными тремя связями, меняется в пределах от 0 до 1. Для силового поля AMBER необходимо использовать 0.5, для OPLS – 0.125, для BIO+ - 1.0.
Cutoffs (Отсечения) определяет расстояние, после которого нековалентные взаимодействия между атомами не учитываются. Его необходимо вводить для того, чтобы избежать учета взаимодействия с соседями по периоду в случае расчетов в Periodic Box.
![]()
![]()
![]()